

Abstract
Introduction:
Congenital TTP (Thrombotic Thrombocytopenic Purpura) is a rare life-threatening inherited disorder, resulted from mutations in ADAMTS13 gene. Such mutations interfere with ADAMTS13 enzyme production or function, subsequently resulting in microangiopathic hemolytic anemia (MAHA) and microvascular thrombosis. The investigation of congenital TTP is challenging because of its rarity with an estimated prevalence ranging from 0.4 to 16.7 cases per million and about 150 identified mutations. Recently, we identified a cohort of patients with congenital TTP from three consanguineous families of Arab Bedouin origin in southern Israel. All patients have the same, previously unidentified mutation c.3772 delA in the ADAMTS13 gene. In this single center prospective cohort study, we present patients' genetic, clinical and laboratory data and results of the abnormal ADAMTS13 protein Western blot analysis.
Methods:
Patients with episodes of MAHA and thrombocytopenia were diagnosed with congenital TTP by abnormally low ADAMTS13 activity and absence of inhibitory autoantibodies (Table 1). Subsequently, patients were referred for genetic testing. Patients' DNA served for PCR amplification of the exons of ADAMTS13 (NM_139026) followed by direct sequencing of the PCR products. The mutation was found in exon 28, the primers used for PCR were forward: atgtccctatgtcccacctg and reverse: ctgtccagaatcacagcacaa (annealing temperature 61°C).
Western blot analysis of the abnormal ADAMTS13 protein was performed according to the method previously described by Zheng et al, using antihuman ADAMTS13 rabbit polyclonal antibody (ab28274; Abcam, Cambridge, UK).
Patients' clinical and laboratory parameters were collected at diagnosis and during follow-up.
The study was approved by the institutional research ethics board.
Results:
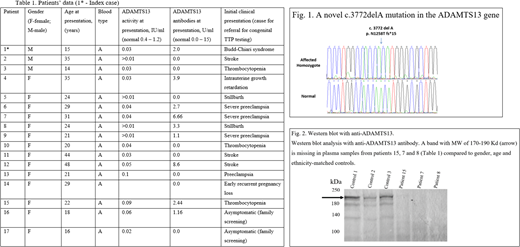
We identified a novel mutation on chromosome 9:136323172del A (GRCh37/hg19) in coding exon 28, c.3772 del A (NM_139026), causing p.N1258T fs Ter 15 of ADAMTS13 gene close to the C-terminus of the 1427-amino acid long protein (Fig.1). We validated that all patients in our cohort are homozygous for the mutation. The mutation is found within the CUB1 domain and the frameshift is predicted to result in the loss of the next 169 highly conserved amino acids of the full-length protein that contains the CUB2 domain, normally responsible for cleavage of large VWF multimers under flow conditions.
Our Western blot analysis of the abnormal protein (Fig. 2) demonstrates that a 170-190 Kd band is missing in plasma of five patients, compared to four controls. These results suggest that the mutation may interfere with the entire enzyme production.
The cohort consists of 17 homozygotic patients (3 males and 14 females), all belong to three closely related families of Arab Bedouin descent with high rate of consanguinity. All 17 patients in this cohort have blood group A. This finding may represent genetic linkage disequilibrium between the ADAMTS13 gene and the transferase gene, both found on chromosome 9 in close proximity.
Despite the fact that all the patients are homozygous for the same mutation and belong to three closely related families, their presenting symptoms and disease severity were markedly heterogenous (Table 1): eight of 14 female patients initially presented with complications of pregnancy, two presented with laboratory abnormalities, two presented with stroke and two asymptomatic female patients were recognized on family screening. Of three male patients, one presented with Budd-Chiari syndrome, one had laboratory abnormalities alone and one developed a stroke.
Conclusion:
To the best of our knowledge, this is one of the largest congenital TTP cohorts described in the literature. This cohort is unique due to the fact that all members carry the same, previously unreported, ADAMTS13 gene mutation in the CUB1 domain of the gene. Our findings support an assumption that environmental and hereditary modifiers may influence disease course. Further research of the involved families may enable us to expand the understanding of the pathophysiology and develop better treatment options for this understudied rare condition.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal